


濟南順奇凈化工程有限公司
電話(傳真):0531-68824415
手 機:13854165330
Q Q:340095748
聯系人:張經理
郵 編:250024
郵 箱:340095748@qq.com
地 址:山東省濟南市天橋區新徐居委會黃河建邦大橋西側1-6號
原料藥一般由化學合成、DNA重組技術、發酵、酶反應或從天然物質提取制得,它是加工藥物制劑的主要原料, 原料藥分無菌原料藥和非無菌原料藥。前者是要按規定進行法定的無菌檢查,后者則不需要作此檢查。
GMP(2010)對非無菌原料藥的精、烘、包操作工序的暴露環境推薦采用無菌藥品的D級區標準。
所謂“精”,即精制,包括精濾、結晶、分離、檢驗等工序。
所謂“烘“,即干燥,包括干燥、粉碎、混粉及檢驗等工序。
所謂“包”,即包裝,包括包裝材料的處理和包裝等工序。
原料藥精、烘、包各工序的特點是:
(1)精濾對法定要無菌檢查的,對無菌藥品確定環境級別;不需無菌檢查的,則只要求D級環境。
(2)結晶要求與精濾相同。
(3)分離注意分離有機溶媲或有氣妹的品種時,應采取措施,防止溶媒或氣味在室內擴散。
(4)干燥(俗稱烘干)盡可能采用干燥、混粉一次完成的設備;干燥時所用的空氣應經凈化處理,達到與生產環境同等潔凈程度;尾氣需經捕集、除塵后再排放。
(5)粉碎和過篩應有局部防塵或吸塵裝置。
(6)檢驗。
(7)包裝材料處理注意直接接觸藥品的包裝材料應按適當方法清潔消毒,其中無菌原料藥所用的內包材要用經過過濾的注射用水沖洗,并在4h內滅菌,24h內使用,且均應專柜貯存在與包裝同等潔凈度級別的貯存室內。
(8)包裝有換批生產時,注意做好清場工作。
圖9-1是非無菌原料藥工藝示意圖。
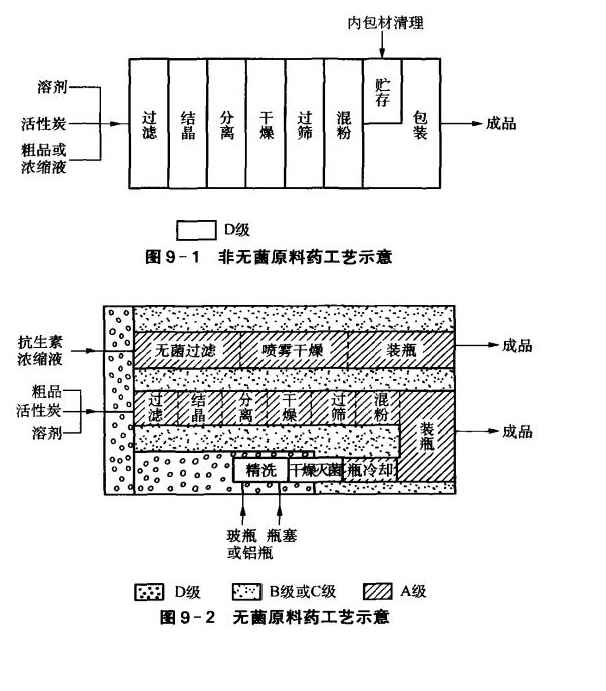
圖9-2是無菌原料藥工藝示意圖。
(9)廠房設計GMP規定原料藥的質量標準中有熱原或細菌內毒素等檢驗項目的,廠房的設計應特別注意防止微生物污染,如應根據工藝要求設定相應廠房的潔凈度級別。
質檢實驗室通常應與生產區分開。當生產操作和檢驗結果之間無互相不利影響時,中間控制實驗室可設在生產區內。
GMP(2010)的術語定義指出中間控制“也稱過程控制,指為確保產品符合有關標準,生產中對工藝過程加以監控,以便在必要時進行調節而做的各項檢查。”當然,中間控制操作不得給藥品帶來質量風險。
(10)設備可用同一設備生產,但應有防止交叉污染的措施,應有適當間隔。難以清潔的設備或部件應專用。在無菌操作條件下添加細胞基質、培養基、緩沖液和氣體,應當采用密閉或封閉系統。
(11)清洗更換批次或品種生產前,必須對設備徹底清洗。
(12)發酵工藝采用發酵工藝生產原料藥應根據生產步驟和生產條件(敞口、密閉或封閉系統)確定環境控制標準。初始容器接種、轉種或加料(培養基、緩沖液)使用敞口容器操作時,如微生物污染可能危及原料藥質量時,敞口容器應置于適當的控制環境中。 需在無菌操作條件下添加細胞基質、培養基、緩沖液和氣體時,應采用密閉或封閉系統。
(13)容器對可重復使用的容器應行清潔并去除或涂毀其原有標簽。
