


濟南順奇凈化工程有限公司
電話(傳真):0531-68824415
手 機:13854165330
Q Q:340095748
聯系人:張經理
郵 編:250024
郵 箱:340095748@qq.com
地 址:山東省濟南市天橋區新徐居委會黃河建邦大橋西側1-6號
在
關于這兩種看似不同的模式,歐盟 GMP 指南(2009)引言中有一段耐人尋味的表述:“本指南無意成為任何新概念或新技術發展的障礙,如果通過驗證并證明所用方法能達到至少與本指南所述方法等價的質量保證水平,也應予以認同(TheGuide is not intended to place any restraint upon thedevelopment of any new concepts or new technologieswhich have been validated and which provide a level of Quality Assurance at least equivalent to those set out in this Guide)”。同時,它還明確表達“藥品多年來按 GMP 要求生產,沒有執行 ISO 的標準。企業可自行決定采用ISO標準作為實施制藥領域質量體系的一種手段(The manufacture of medicinal products has for many years taken place in accordance withguidelines for GMP and the manufacture of medicinalproducts is not implement by ISO standards. Harmo-nized standards as adopted by the ISO may be used atindustry’s discretion as a tool for implementing a quality system in the pharmaceutical sector)”。
同樣,FDA 的指南文件也有類似內容的表述,足見這兩種模式在國際制藥行業的互認和共容。歐盟GMP指南與ISO14644.4 在控制方法上的差異,并不說明它們在控制水平上的差距。國際制藥企業在實施時可自行選用,無須厚此薄彼,只要認真執行都能達到同樣的質量保證水平。因此,美國制藥企業產品無需按歐盟的ABCD模式方可進入歐盟國家;同樣歐盟制藥企業產品也沒有必要改成FDA 的ACD 模式才能進入美國市場。可是,當這兩項國際標準演變成我國的法規和標準時,我國的制藥企業就沒有這么幸運。因為我國新版GMP在等效采用歐盟GMP指南時,刪除了歐盟GMP指南的這段“提示”。新版 GMP 發布前,我國以往 GMP中無菌藥品生產環境的空氣潔凈度標準基本采用ACD 模式。客觀上,我國歷版 GMP 內容和企業執行力度都與國際要求存在一定差距,追其原因錯綜復雜,我國無論在認知理念、綜合國力、技術水平等方面都無法與國外發達國家相比,即使照搬照套國外 GMP,也不能起到立竿見影的效果。如果新版 GMP 能根據國情吸納歐盟 GMP 指南的寬容,大多數企業可在原有基礎上進行填平補齊的改造,而無需如今傷筋動骨式的重建。這無論對國家還是對企業,都是值得深思的大事。
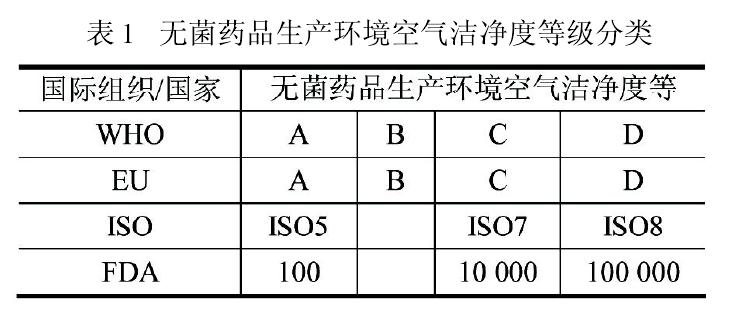
新版 GMP 是強制執行的國家行政法規,是制藥企業認證檢查的唯一標準,得不到認證證書,企業必須關停并轉;而B/T25915 是國家推薦標準,又無相應檢查頒證手段,自然無法與新版GMP相提并論。出現如此問題,并不說明這兩項標準的水平差距,而是國家行政制度、權力上的失衡,需要由政府有關部門采取措施予以彌合修補。GB/T 25915.4(2010)-設計、建造、啟動,即 ISO14644.4 的附錄 B,提出了無菌產品在顆粒物和微生物受控的潔凈區內無菌灌裝線上灌裝的空氣潔凈度等級要求(表 2 )。
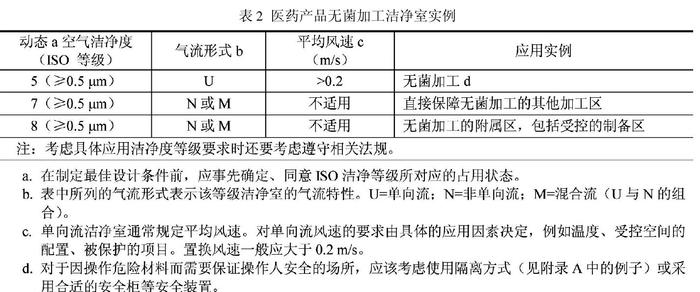
表2中值得我們注意的 ,一是空氣潔凈度等級的占有狀態應在設計前由業主和設計人員事先商議確定。這是 ISO14644 系列標準在定義“動態”、“靜態”時的一貫主張,因為各種占用狀態都有它的使用價值,不能簡單地以使用何種“占用狀態”來評判標準的孰高孰低。受無菌藥品灌裝線上人員操作、設備性能、生產運行等因素影響,單向流罩下的空氣潔凈度往往難以測準,為確保測試狀態的穩定性和判斷結果的可比性,日本把測試要求原則上定為“靜態”,也有國家規定動態檢測范圍不包括操作點或發塵點
GB/T25915.4(2010)有著與新版 GMP 同樣的控制理念,都為了有效控制藥品生產環境的污染。圖 1 是 GB/T25915.2010)的層式污染控制概念。工藝核心區(與環境發生交互作用的工藝位置)所在的區域為潔凈區,是潔凈室中受控最嚴的部分,空氣潔凈度等級為單向流100 級(即ISO 5 級,歐盟 A 級)。出于經濟、技術和運行等方面的原因,要盡量減小最高潔凈度區域的范圍。潔凈區通常采用密閉式(如 RABS),或由潔凈度較低的外部區域-潔凈室(1 萬級,即 ISO7 級)包圍。相鄰區域間的人流、物流進入工藝核心區時,會增加傳播污染的風險,因此應特別注意人流和物流的布局細節與管理,這就是層式污染控制的基本概念。
與 GB/T25915.4(2010)不同的是新版GMP將包圍潔凈區的外部區域(潔凈室)的空氣潔凈度等級設定為B級,所謂B級是靜態控制時相同于A 級(單向流 100 級)的靜態,動態控制時相同于 C 級(1 萬級)的靜態。GB/T 25915.4(2010)和新版 GMP 都認為無菌灌裝線外部區域(潔凈室)空氣潔凈度等級應低于工藝核心區(潔凈區),至于低多少,是1 萬級還是B 級?這是可探討的學術問題,不應成為判定標準高低的分界線。國內外實踐證明,背景設置為1 萬級或B 級,都能保護無菌藥品的生產環境,我們應從分析影響無菌灌裝線的環境因素著手,尋找控制污染源的途徑,才能最終確保無菌生產環境。
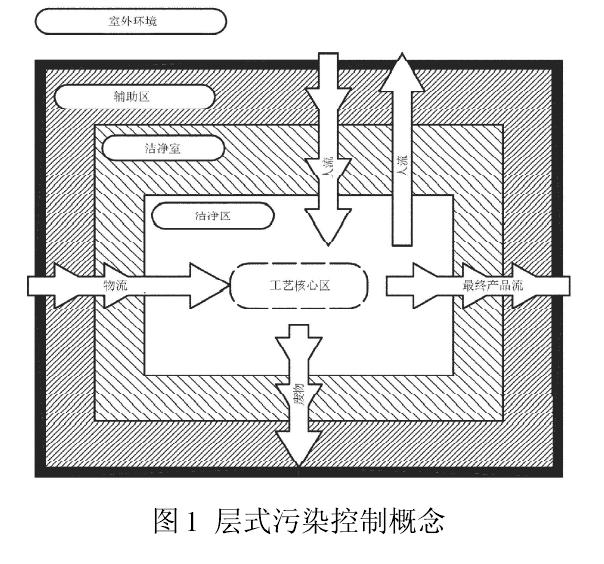
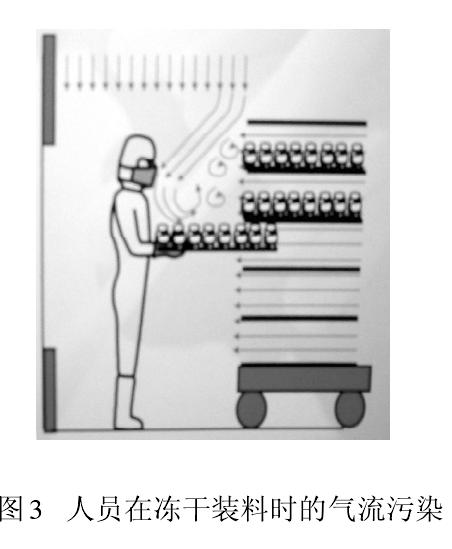
從人們開始接受并應用潔凈技術時就發現,人是潔凈室最大的污染源。以微粒和微生物為污染物控制對象的制藥生產,大量測試數據和生產實踐告誡我們,人員(生產、檢驗、管理、維修)因素無時無刻不在干擾藥品生產。圖2 顯示人員在單向流下操作導致氣流粒子污染。此刻,保護性的單向流變成污染物的載體,直接影響被保護的產品。此時氣流速度越大,則污染程度越大。圖3 為凍干劑裝料時,單向流罩下垂直和水平氣流相互沖突,
形成紊流,紊流氣流會在較長時間里產生懸浮污染。
不言而喻,人是無菌灌裝過程中影響最大的環境因素。為此,實現無菌灌裝環境控制的技術要點是:(1)將人員從無菌環境隔離,從根本上排除人的干擾;(2)采取局部隔離無菌環境 (如R A B S 等);(3 )即便如此仍需限制人員對無菌產品的干預;(4)生產運行中不讓人員進入無菌環境。不解決人對無菌環境干擾問題,大幅度提高無菌藥品生產線外部區域空氣潔凈度等級,效果也不會明顯。
必須強調,醫藥工程設計中空氣凈化措施只是控制潔凈室微生物的手段之一,這些措施只能控制通過空氣途徑傳播的微生物污染,對表面對象微生物的生物污染以及化學污染都無能為力,藥品生產使用的設備、設施、容器具、物料、包裝材料等沾附的微生物、化學品,以及生產、管理人員攜帶的微生物,只有采用清場、清洗、消毒、滅菌等措施,任何級別的凈化空調都無法替代。
近年來國外對空氣凈化的濾菌效果提出質疑。數據表明 1 臺
凈技術老路,將會大大浪費潔凈室建設費用和運行成 本 。
國標 GB/T 25915.4(2010)的內容包括規劃和設計、建造和啟動、檢測和驗收、文件等方面要求,涵蓋工程建設的全過程,對我國醫藥行業尤為重要。藥品生產實施 GMP 是一項系統工程,為藥品生產提供符合 GMP 要求的廠房設施是企業實施G M P 的先決條件。因此,醫藥工程項目必須按GMP 要求實施規范管理,才能為企業實施 GMP提供可靠保障。近年來國外針對醫藥工程項目的規范化管理,一項新的管理規范― ― GEP已在一些發達
國家悄然問世。GEP(醫藥工程質量管理規范)是Good Engineering Practice 的縮寫,是與GMP配套的藥品生產管理文件。它的宗旨是“將已確立的工程設計方案和標準應用于項目的整個生命周期中,提供合適成本的解決方案”。我國至今尚無這方面的規范,工程項目大多由企業憑經驗自行管理,主要依據是各類相關的國家標準,缺乏系統性、協調性。
國標 GB/T 25915.4(2010)的發布,為工程項目規劃、設計、施工、試車、檢測、驗收等環節的規范化管理填補了空白,國標所附的大量資料性附錄,如“設施的驗收”、“設施的布局”、“建造和材料”、“潔凈室的環境控制”、“待需方/ 用戶與供方/ 設計方商定的補充技術要求”等,為項目的工程設計、采購供應、施工安裝、試車驗收、工程進度、技術質量、安全環保、資金運作、部門協調等提供了詳盡的規范要求,制藥企業應自覺以國標GB/T 25915為準繩,并據此制訂醫藥工程項目的GEP,確保工程項目符合 GMP 要求。
國標GB/T 25915系列標準反映了當今國際“潔凈室及相關受控環境 ”的新概念、新思路和新方法,是指導我國各行各業潔凈技術的規范性文件,必將成為我國潔凈技術蓬勃發展的新起點。
本文標簽:潔凈室
